
Marine Metagenomics for New Biotechnological Applications


Concept
MAMBA stands for “Marine Metagenomics for New Biotechnological Applications”, a collaborative project to mine for and use of new microbial activities, in particular for targeted production of fine chemicals, antioxidants and anti-cancer drugs.
This Project builds up on the previous efforts of European Framework Programs and National research initiatives for exploiting the catalytic activities of marine microorganisms and microbial communities and to gain the new knowledge on the mechanisms of survival of living organisms in extreme environments.
The urge for the integrated approach in metagenomics
The majority of current biotechnological applications are of microbial origin, and it is widely appreciated that the microbial world contains by far the greatest fraction of biodiversity in the biosphere, so it is the microbes that will deliver the greater part of enzyme diversity and the majority of new applications. Because it is generally accepted that marine microbial communities account for more than 80% of life on Earth, and have an indispensable role in primary energy and carbon recycling, marine biochemical and chemo-diversity is considered to be the major target for the prospecting for new enzymes and natural products, e.g. for drug development.
However, the well-known dilemma of microbes – that the majority cannot be cultivated – limits application of the traditional means of enzyme discovery described above.
The anticipated rich enzymatic pickings from the uncultured microbial majority has stimulated the development of new genomics-based discovery approaches, the so-called “metagenomics” or environmental genomics approaches. There are two distinct strategies taken in metagenomics, according to the primary goal. A large-scale sequencing of either bulk DNA through pyrosequencing techniques, or sequencing the DNA libraries constructed for archiving and sequence homology screening purposes, is aiming at capturing the largest amount of the available genetic resources present in the sample or archive for its further data mining, mostly homology-based. In contrast to that, small insert expression libraries, especially those made in lambda phage vectors, are constructed and implemented for a direct activity screening. The clear disadvantage of the first approach is its full reliance on the existing genome annotation data that (1) limit the prediction protein/enzyme to already known protein families and thus do not allow to discover really new proteins with novel functions, or (2) these genome annotation data are simply erroneous.
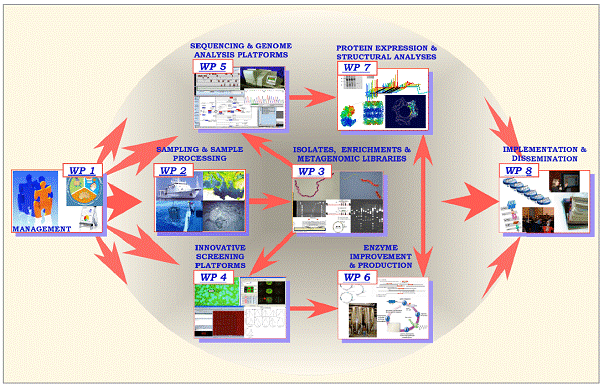
Unfortunately, the generation of vast metagenome sequencing data is not followed, at same extent, by the discovery of new activities, or functional characterisation of proteins from the plethora of genes from these metagenomes. To cope with this, a number of structural genomics groups were established worldwide, whose ultimate goal is to obtain the structures for at least one protein representing each protein family of 10.000-15.000 currently known. However, the majority of the protein structures does not suggest the function of these proteins! The reasonable answer to this challenge will be to combine the en masse activity characterisation of the proteins with elucidation of their structures and with the bioinformatics approaches and consequent channelling these new activities towards the new biotechnological applications.